分子化学工学
新田研究室では、分子シミュレーションと量子化学計算を用いて相平衡・吸着・膜透過・化学反応機構などの研究を行っています。研究手段がコンピュータを用いた理論計算だけであるという点では、日本(世界)の化学工学の中で数少ない研究グループの一つです。そこで、コンピュータシミュレーションの背景を説明し、これからの展望を述べましょう。
1.コンピュータによる計算グループ
実験と理論は車の両輪に例えられます。自然現象を理解し自然に働きかけるには、この両輪が必要で、実験も理論もできることが理想です。しかし、理論が精密になり、大規模な理論計算に必要な計算機数学やプログラミングに関して必要とされる知識が広がるにつれて、理論計算の仕事が専門化してきました。分子シミュレーションや量子化学計算の研究最前線はそのような分野になっており、私たちも相平衡・表面現象や反応機構の研究分野で専門化した計算グループの一つです。
2.コンピュータシミュレーション:その利点と問題点
計算機シミュレーションの第一の利点は、実験が難しい条件(高温・高圧・低温などの極限条件)での計算が可能なことでしょう。この計算には、分子の大きさだけを変えるとか、ポテンシャル深さだけを変えるような場合も含まれます。これによって、着目する現象を支配する主要な因子を正確に調べることができます。
第二の利点は、実験で求めることがもともと無理な情報(局所密度分布、波動関数など)を得ることができます。この情報の生かし方については、3次元グラフィックスやアニメーション技術が特に有効になります。
第三の利点は、計算に必要な理論(古典力学、統計力学、量子力学など)の基本原理を深く理解できることです。これを第一の利点というべきかもしれません。実際、原理を正確に理解しないと正しい計算結果は得られず、計算された結果の正しい解釈もできないのです。
最後は希望的な見通しですが、実験値との一致が保証されているパラメータの適用範囲を注意して使えば、特定の機能を持った材料や分子の設計が計算だけで可能になるでしょう。コンピュータ支援による材料設計・分子設計への展望です。
分子シミュレーションの問題点は、分子間ポテンシャルに関する知識・データベースがまだ不足していることです。また、第一原理量子化学計算は長い計算時間が必要なので、現在の計算機の性能では系が小さく限定されます。しかし、計算の並列化手法や量子化学計算と古典力学との折衷・ハイブリッド(QM/MM)法の導入など、新しい手法の導入と計算機のさらなる性能アップが見込める現在、若い人たちが活躍できる場が広がっています。
3.計算方法とプログラム
計算方法は研究対象に応じて変わりますが、相分離・吸着等温線などの熱力学的な物性の研究にはモンテカルロ(MC)法を、細孔内拡散や膜透過現象の研究には分子動力学(MD)法を、ラジカルやイオンの関与する反応機構の研究には第一原理分子動力学(ab
initio MD)を用いています。研究で使うプログラムは手作りを基本としていますが、Gaussian、Gamess、Crystalなどの量子化学計算ソフトとAVSなどのアニメーション作成ソフトは市販品を使っています。
4.これまでの研究とこれからの研究
これまで取り上げてきた研究テーマを対象で分類すると、超臨界流体・吸着・膜透過・表面修飾・固相の相分離・ラジカル/イオン反応機構などに分けることができます。
(1)超臨界流体(SCF)は、通常の気体および液体とは違った特異な性質を持つので、新しい分離精製プロセスや反応プロセスの開発に応用できることが期待されています。SCF中の吸着に関する研究では、MC計算を組み合わせることにより、SCFの持つ特異な溶解力と固体表面の吸着能および溶質とSCFの競争吸着という3つの因子によって吸着特性が決まることを明らかにしました。
(2)超臨界流体を急速に膨張させると、溶解していた物質を微粒子や糸状で取り出すことができるという報告があります。最近、SCFの断熱膨張過程をシミュレーションしましたが、そのスナップショットの一例を示します。
 急速断熱膨張過程のシミュレーション
急速断熱膨張過程のシミュレーション
(3)密度勾配がある系のシミュレーションができる境界制御型の非平衡MD法を開発しました。これを使って気体の無機膜の透過現象を研究し、透過抵抗に対する気体の吸着力と細孔内拡散速度の寄与について報告しました。モデル化した炭素膜の中を透過する気体分子のスナップショットの一例を示します。膜の構造と膜の分離特性については、透過する混合気体の競争吸着と相対的拡散速度が効いていることは確かですが、膜の入口・出口の分離機能に関しては既存の理論が見つからないという結果が得られています。
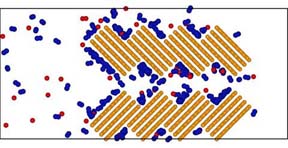 炭素膜の透過シミュレーション
炭素膜の透過シミュレーション
(4)吸着剤や膜剤の表面修飾によって特定物質の吸着力を大きく(小さく)するような表面修飾法が、第一原理量子化学計算によって可能かどうかを研究しています。二酸化チタンの表面に吸着したアンモニア分子の計算図を示します。この研究は分子設計戦略への一つの試みで、これからが楽しみです。
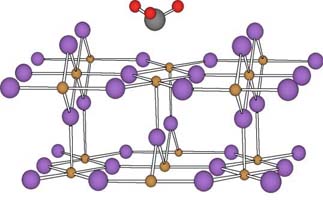 TiO2-NH3系の量子化学計算
TiO2-NH3系の量子化学計算
(5)超臨界水中での反応がイオン的に進むのか、ラジカル的に進むのかについて、論争があり、決着がついていません。実空間差分法を用いた第一原理分子動力学法を新規に開発して、OHラジカル/OHイオンとホルムアルデヒドあるいは塩化メチルの反応を研究しています。塩化メチルの反応ではいわゆるSN2反応の過程を見ることができます。さらに、ハイブリッド(QM/MM)法のプログラムができたので、反応機構に及ぼす溶媒和の効果が直接計算できるようになりました。図は、エタノール酸化反応のQM/MM計算のスナップショットです。
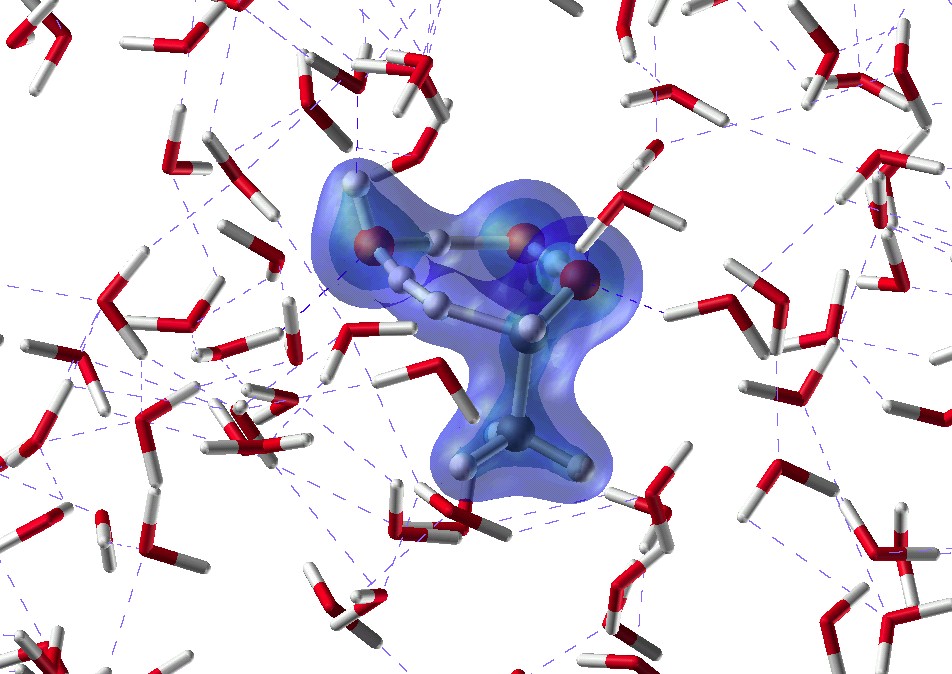 反応経路の追跡
反応経路の追跡
(6)溶融したガラスを急冷すると、成分組成の偏った固体のガラスができます。これをスピノーダル分解といいます。アルカリ成分であれば酸洗浄によって酸領域を除去することができ、除去後が細孔になります。細孔径を制御するために、ガラスの組成・急冷速度と細孔径の関係を知りたいのです。このような観点でのシミュレーションを計画しています。
駆け足で研究内容を紹介しました。詳しくは最近の論文や研究発表をご覧ください。
Recent publications
Shin-ichi Furukawa, Keigo Goda, Yi Zhang and Tomoshige Nitta: Molecular Simulation Study on Adsorption and Diffusion Behavior of Ethanol/Water Molecules in NaA Zeolite Crystal, J. Chem. Eng. Japan, in press
Shin-ichi Furukawa, Satoshi Kato and Tomoshige Nitta: Molecular Dynamics Studies on Clustering Process of Solute Molecules through Rapid Expansion of Supercritical Fluids, Fluid Phase Equil., in press
Takumi Hori, Hideaki Takahashi, and Tomoshige Nitta: Hybrid quantum chemical studies for the methanol formation reaction assisted by the proton transfer mechanism in supercritical water; CH3Cl + nH2O -> CH3OH + HCl + (n-1) H2O, Journal of Chemical Physics, 119(16), 8492-8499 (2003)
Hideaki Takahashi, Hideaki Hashimoto, and Tomoshige Nitta: Quantum mechanical/molecular mechanical studies of a novel reaction catalyzed by proton transfers in ambient and supercritical states of water, Journal Chemical Physics, 119(15), 7964-7971 (2003)
Yi Zhang, Shin-ichi Furukawa and Tomoshige Nitta: Molecular Simulation Studies on Adsorption of Propane/Propylene in NaA zeolite by Using a Monte Carlo Technique, J. Chem. Eng. Japan, 36(9), 1085-1094 (2003)
Hideaki Takahashi, Shigeki Takei, Takumi Hori, and Tomoshige Nitta: A real-space-grid QM/MM study on the ionic/radical association reaction in aqueous phase: HCHO + OH -> HCHO-OH, Journal of Molecular Structure (Theochem), 632, 185-195 (2003)
Yi Zhang, Shin-ichi Furukawa and Tomoshige Nitta: Computer Simulation Studies on Gas Permeation of Propane and Propylene across ZSM-5 Membranes by a Non-Equilibrium Molecular Dynamics Technique, Separation and Purification Technology, 32(1-3), 215-221 (2003)
Shin-ichi Furukawa and Tomoshige Nitta: A Study of Permeation of n-Butane in ZSM-5 by using Monte Carlo and Equilibrium/Non-Equilibrium Molecular Dynamics Simulations, J. Chem. Eng. Japan, 36(3), 313-321 (2003)
Hori T., H. Takahashi, and T. Nitta: Hybrid QM/MM Molecular Dynamics Simulations for an Ionic SN2 Reaction in the Supercritical Water: OH- + CH3Cl → CH3OH + Cl-, J. Computational Chem. 24(12), 209-221 (2003)
堀 拓実・高橋英明・新田友茂; 実空間差分法による非周期系第一原理分子動力学法の開発とその応用-塩化メチルと水酸化物イオンのSN2反応-, JCPE Journal, 13(1), 21-28 (2001)
Hideaki Takahashi, Shumpei Hisaoka, Tomoshige Nitta: Ethanol Oxidation Reactions Catalyzed by Water Molecules: CH3CH2OH + nH2O → CH3CHO + H2 + H2O (n=0,1,2), Chem. Phys. Lett., 363(12), 80-86 (2002)
Chong Gu, Guang-Hua Gao, Yang-Xin Yu and Tomoshige Nitta: Simulation for Separation of Hydrogen and Carbon Monoxide by Adsorption on Single-Walled Carbon Nanotubes, Fluid Phase Equil., 194-197, 297-307 (2002)
Furukawa, S., C. McCabe, P.T. Cummings, and T. Nitta: Non-equilibrium Molecular Dynamics Simulation Studies on Behavior of Hydrocarbon-isomers in Silicalite, Fluid Phase Equil., 194-197, 309-317 (2002)
Takahashi, H., K. Yuki, and T. Nitta: Chemical modification of rutile TiO2 (110) surface by ab initio calculations for the purpose of CO2 adsorption, Fluid Phase Equil., 194-197, 153-160 (2002)
Takahashi H., T. Hori, T. Wakabayashi and T. Nitta: Ab initio Molecular Dynamics Simulations of an OH Radical / OH Anion with a Solvated Formaldehyde Molecule, Foundations of Molecular Modeling and Simulation, AIChE Symp. Ser. No. 325, vol. 97, 280-282 (2001)
Takahashi, H., T. Hori, H. Hashimoto and T. Nitta: A Hybrid QM/MM Method Employing Real Space Grids for QM Water in the TIP4P Water Solvents, J. Computational Chem., 22(12), 1252-1261 (2001)
Takahashi, H., T. Hori, T. Wakabayashi, and T. Nitta: Real Space Ab Initio Molecular Dynamics Simulations for the Reactions of OH Radical/OH Anion with Formaldehyde, J. Phys. Chem. A, 105(17), 4351-4358 (2001)
Nitta, T. and S. Furukawa: Simulation Performance of a Non-Equilibrium Molecular Dynamics Method using Density Difference as Driving Force, Molecular Simulation, 25, 197-208 (2000)
Furukawa, S. and T. Nitta: Non-equilibrium molecular dynamics simulation studies on gas permeation through carbon membranes with different pore shape composed of micro-graphite crystallites, J. Membrane Sci., 178, 107-119 (2000)
Takahashi, H., T. Hori, T. Wakabayashi and T. Nitta; A Density Functional Study for Hydrogen Bond Energy by Employing Real Space Grids, Chemistry Letters, No.3, 222-223 (2000)
Takahashi, T., K. Okuda and T. Nitta; Chemical Modification of MgO (001) Surface by Utilizing Energy Decomposition Analyses for the Purpose of CO2 Adsorption, Bull. Chem. Soc. Jpn., 73, 315-319 (2000)
Furukawa, S., T. Sugahara and T. Nitta; Non-equilibrium MD Studies on Gas Permeation through Carbon Membranes with Belt-like Heterogeneous Surfaces, J. Chem. Eng. Japan, 32(2), 223-228 (1999)
Shigeta, T. and T. Nitta; Monte Carlo Simulation Study of Entrainer Effect on Distribution Equilibrium between Supercritical Fluids and Slitpores, J. Chem. Eng. Japan, 32(1),150-152 (1999)
Takahashi, H., T. Wakabayashi and T. Nitta; A wavepacket analysis of the statistical behaviour of molecules that penetrate a slit-shaped micropore, Chem. Phys. Letters, 294, 95-102 (1998)
Nitta, T. and T. Shigeta; Computer Simulation Studies of Adsorption Characteristics in Supercritical Fluids, Fluid Phase Equilibria, 144(1-2), 245-256 (1998)
Takahashi, H., Nakamura, and T. Nitta; Effect of the Shape of Simulation Box on the van der Waals loop of Lennard-Jones Fluid, Chem. Phys. Letters, 282, 128-132 (1998)
Furukawa, S., K. Hayashi and T. Nitta; Effects of Surface Heterogeneity on Gas Permeation through Slitlike Carbon Membranes by Non-Equilibrium Molecular Dynamics Simulations, J. Chem. Eng. Japan, 30(6), 1107-1112 (1997)
Furukawa, S. and T. Nitta: Computer Simulation Studies on Gas Permeation through Nanoporous Carbon Membranes by Non-Equilibrium Molecular Dynamics, J. Chem. Eng. Japan, 30(1), 116-122 (1997)
Nitta, T., H. Kiriyama and T. Shigeta; Monte Carlo Simulation Study for Adsorption of Dimers on Random Heterogeneous Surface, Langmuir, 13(5), 903-908 (1997)
Furukawa, S., T. Shigeta and T. Nitta: Non-Equilibrium Molecular Dynamics for Simulating Permeation of Gas Mixtures through Nanoporous Carbon Membranes, J. Chem. Eng. Japan, 29(4), 725-728 (1996)
トップページに戻る